深入理解有机共轭分子的发光机制意义重大,这为分子结构的高效设计和应用性能的提升与拓展提供重要的理论指导。近日,我校化学与分子工程学院、费林加诺贝尔奖科学家联合研究中心田禾院士团队在振动诱导发光(VIE)机制的研究上再次取得重要进展,相关研究成果以“Interplay of Steric Effects and Aromaticity Reversals to Expand the Structural/Electronic Responses of Dihydrophenazines”为题发表在《美国化学会志》。
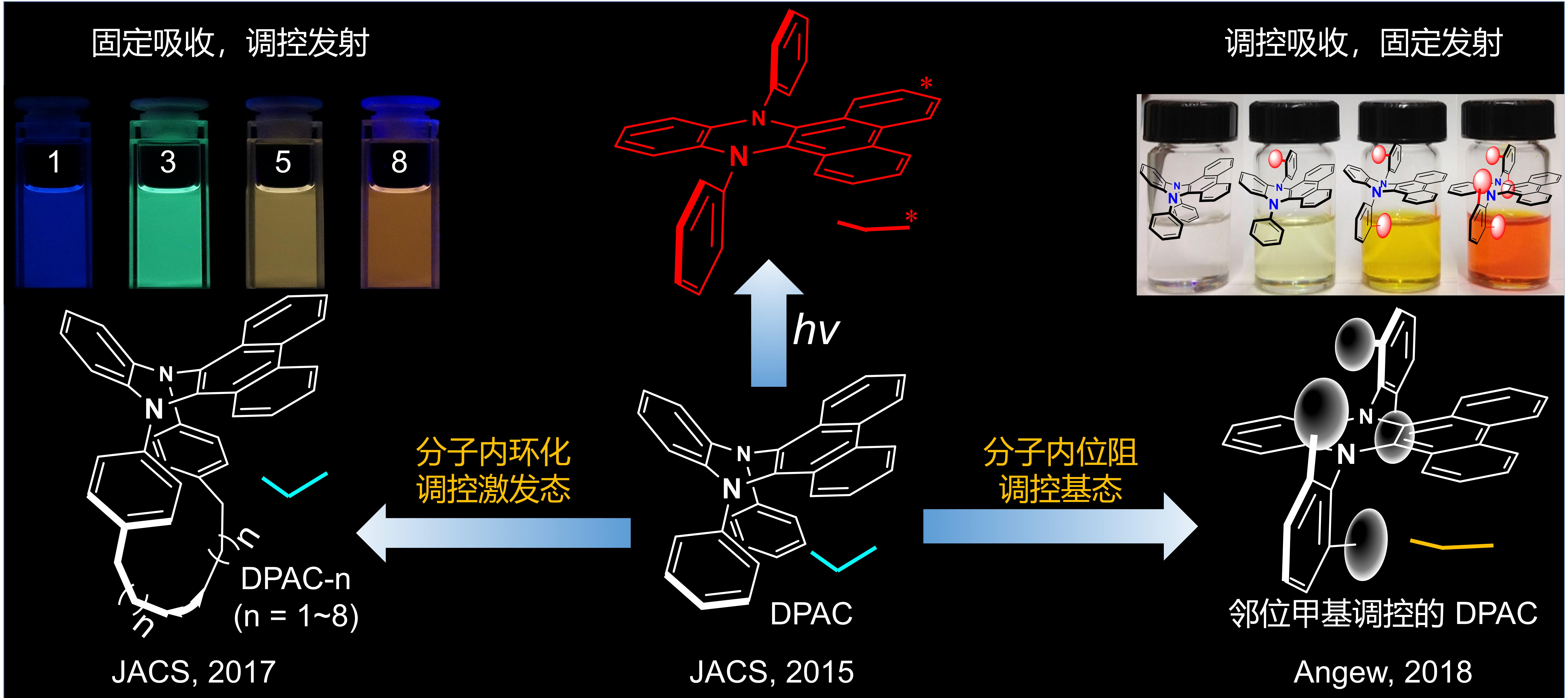
近年来,田禾院士团队在共轭分子发光机制领域做出了独特性和系统性的工作。该团队在研究二氢吩嗪类分子的空穴传输性能时,发现其具有极大的Stokes位移并表现出多重发射的光物理现象,由此提出振动诱导发光(VIE)机制,并通过极性、粘度及温度相关的稳态和超快分辨光谱和飞秒超快光谱研究验证了该理论(J. Am. Chem. Soc., 2015, 137, 8509)。为进一步揭示共轭结构与发光性质之间的相关性,利用“分子内环化调控激发态”的策略实现了对激发态分子骨架演变过程的精准追踪和精细调控(J. Am. Chem. Soc., 2017, 139, 1636);利用“邻位甲基位阻效应”实现了对基态分子构型的逐步精细调控,从而在基态模拟了激发态弯曲到平面化的构象演变过程(Angew. Chem. Int. Ed., 2018, 57, 9880)。在受邀综述中(Chem. Sci., 2020, 11, 7525),田禾院士团队进一步归纳总结出立体位阻和芳香性规则对二氢吩嗪的分子/电子结构和光物理性质起着决定性的作用。
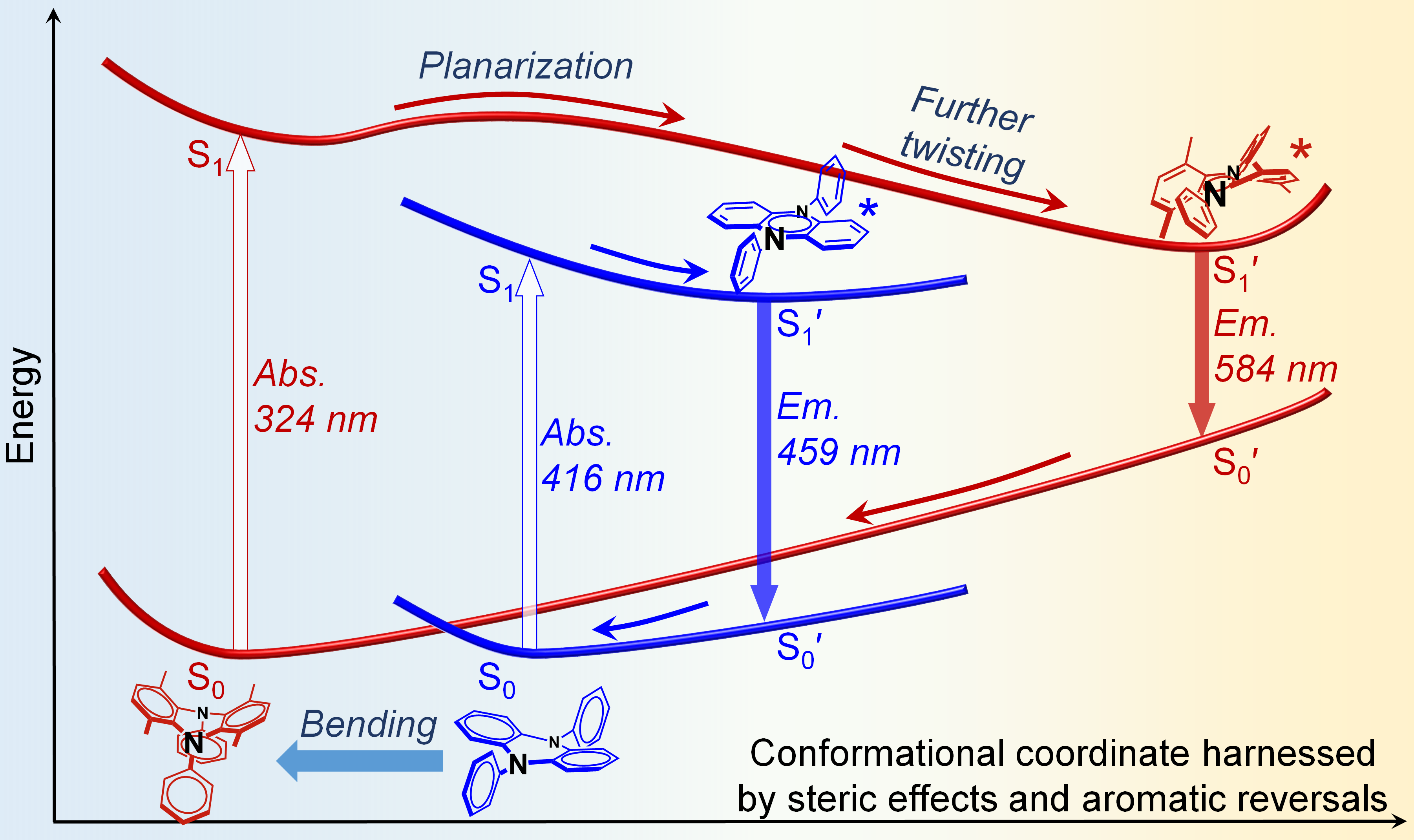
为进一步全面系统地揭示“位阻与芳香性的协同作用”,该研究团队从母体化合物N,N'-二苯基二氢吩嗪入手,在稠环位阻点1,4,6,9-位引入不同位置和数目的甲基,得到一系列不同构型的衍生物,并表现出吸收显著蓝移和发射显著红移的反常光谱行为。理论计算表明,随着甲基数目增加,立体位阻越大,基态分子最优化构象越弯曲使得吸收蓝移,而激发态分子最优化构象越扭曲使得发射红移。其中芳香性翻转是二氢吩嗪稠环结构在基态和激发态构象变化的初始驱动力,而分子内位阻进一步放大了基态弯曲与激发态扭曲的构象差异性,二者的协同作用导致了二氢吩嗪分子独特的基态/激发态分子形变和构象依赖光物理性质。该工作从一个全新的角度全面解读了位阻效应与芳香性规则对共轭分子构型和光物理性质的影响,为指导超大Stokes位移和宽光谱动态发光材料的创制奠定了理论基础和技术支撑。
该工作主要由我校化学院博士生金鑫和李思凡在张志云特聘研究员的指导下完成,并得到了田禾院士和苏建华教授的悉心指导,以及曲大辉教授和花建丽教授的大力支持。计算化学方面得到了复旦大学徐昕教授的指导,超快光谱方面得到了华东师范大学陈缙泉教授、中科院大连化学物理研究所金盛烨研究员的帮助。该工作得到了国家自然科学基金、材料生物学与动态化学教育部前沿科学中心、上海市科技重大专项、上海科学技术委员会、费林加诺贝尔奖科学家联合研究中心等资金支持。
原文链接:https://pubs.acs.org/doi/10.1021/jacs.1c12610